Detecting CRISPR edits
Overview
CRISPR-dependent DNA alterations can sometimes be identified as phenotypic changes in cells. Also, the T7EI assay can be used as a quick assessment of genome editing success, and Sanger sequencing can determine on-target editing. However, next generation sequencing provides much higher resolution necessary to best characterize editing events.

How to confirm CRISPR gene editing success
At the end of a CRISPR genome editing experimental workflow, it is important to determine if the intended edits were made and if there are unwanted off-target edits at other genomics locations. The new genomic sequence at the target site can be characterized using various methods.
The CRISPR basics handbook
Everything you need to know about CRISPR, from A to Z, from theory to practice, for beginners as well as advanced users.
Phenotypic changes
The simplest but often the least specific method of identifying successful CRISPR genome editing is to observe phenotypes of edited cells. In some experiments, the expected phenotype from the gene editing process is known. For example, expression of a fluorescent protein may be turned on or off. In these cases, simply observing phenotypical changes will indicate if genome editing has been successful. However, if sequence information is needed, other assays must be performed as described below.
Enzymatic mismatch cleavage (gel-based) assays
Another simple method to identify on-target gene edits is to use enzymatic assays followed by gel analysis. The T7EI enzyme is used for this purpose. T7EI does not show the actual sequence composition of the edited region but can indicate that a change in the genomic DNA sequence has occurred. This assay is an enzymatic mismatch cleavage method and is available from IDT as the Alt-R™ Genome Editing Detection Kit (the T7EI assay, Figure 1).
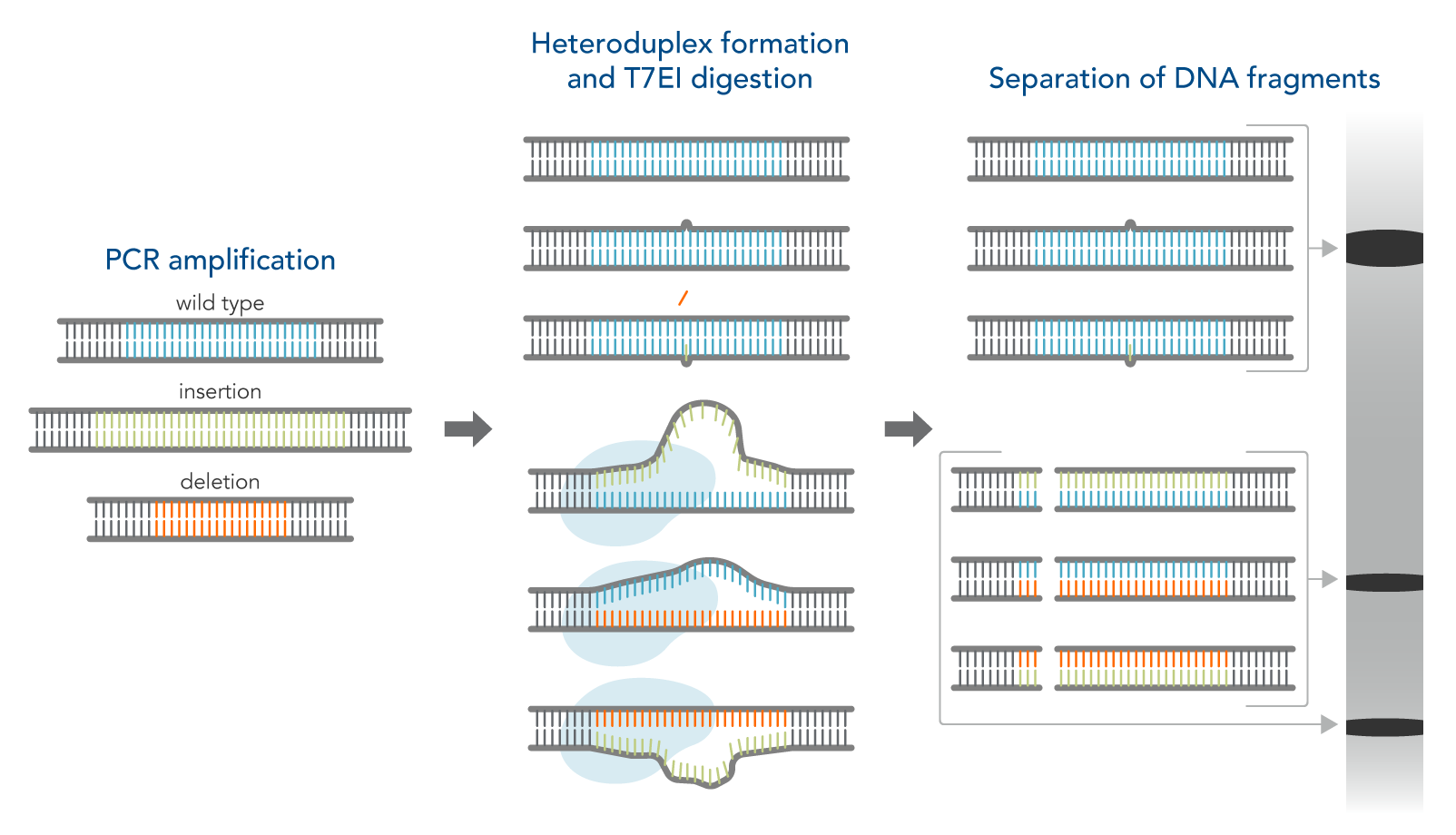
Figure 1. Schematic of T7EI assay.
The workflow for this kit is as follows:
- Step 1—PCR amplification of control and CRISPR-edited genomic DNA. This step increases the amount of available DNA.
- Step 2—DNA duplex/heteroduplex formation. This step combines the original sequence with an edited sequence, resulting in a heteroduplex with mismatched bases at the edited location.
- Step 3—Treatment with nuclease which cuts mismatched DNA.
- Step 4—Analysis of DNA fragments. In this step, the digestion products are run on a gel. The mismatched DNA shows different sizes of bands compared to fully matched DNA.
While the T7EI assay kit (Alt-R Genome Editing Detection Kit) provides a simple method for quantification, it does not accurately identify single-nucleotide changes [1]. Therefore, when using enzymatic mismatch cleavage for quick confirmation of edits, we recommend following up with NGS. NGS provides precise information about the nucleotide changes that have been generated by CRISPR genome editing and can be used to identify off-target effects as well; such detail is not possible with an enzymatic method.
Sequencing methods
Traditional Sanger sequencing was originally designed to identify a single nucleotide sequence, not mixed sequences in variably-edited samples. Therefore, Sanger sequencing is not practical for identification of CRISPR gene edits unless researchers either undertake prior separation of edited DNA (for example, separating cells into pure clones) or use specialized software for deconvolution of sequence data to identify several sequences at a single site. Both cloning and software approaches have been successful for use with Sanger sequencing, but like the enzymatic mismatch cleavage approach, no off-target data can be obtained except within the sequencing read.
Next generation sequencing
NGS is the recommended method for full investigation of CRISPR edits. Highly accurate and consistent, NGS allows identification of even small numbers of edits at both the target site and at nominated off-target sites. The standard approach recommended by IDT scientists is first to nominate off-target sites and then to use the rhAmpSeq™ CRISPR Analysis System to characterize on- and off-target editing. The system depends on IDT’s proprietary rhAmp PCR technology to generate amplicon libraries for targeted sequencing on Illumina® NGS platforms. The system also includes an advanced but accessible cloud-based data analysis pipeline (rhAmpSeq CRISPR Analysis Tool) for quantification of on- and off-target edits. [2]