Site-directed mutagenesis is an in vitro method for creating a specific mutation in a known sequence. While often performed using PCR-based methods, the availability of custom-designed, synthetic, double-stranded DNA (dsDNA) fragments can drastically reduce the time and steps required to obtain the same sequence changes.
Here, we describe several PCR-based methods for site-directed mutagenesis. Primers designed with mutations can introduce small sequence changes, and primer extension or inverse PCR can be used to achieve longer mutant regions. Using these site-directed mutagenesis techniques allows researchers to investigate the impact of sequence changes or screen various mutants to determine the optimal sequence for addressing the question at hand. The IDT Mutagenesis Application Guide provides more details on these approaches.
Read our follow-up article, Site-directed mutagenesis—improvements to established methods, to learn how to use a simplified, alternative approach for generating similar mutagenesis designs quickly with custom-designed, dsDNA fragments.
Traditional PCR
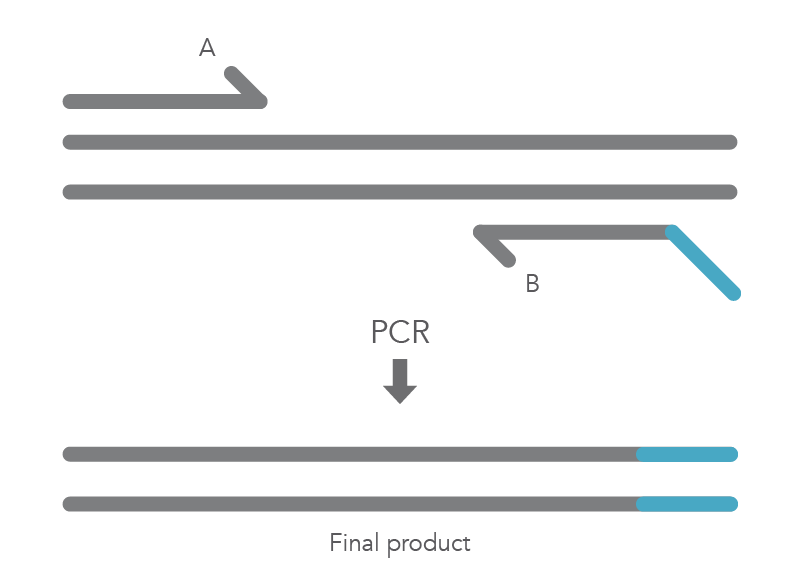
When PCR is used for site-directed mutagenesis, the primers are designed to include the desired change, which could be base substitution, addition, or deletion. The mutation is incorporated into the amplicon during the PCR protocol, replacing the original sequence. Mutations introduced by PCR can only be incorporated into regions of sequence complementary to the primers and not regions between the primers [1]. The PCR protocol for base substitutions involves using primers containing the base changes of interest that occurs as a non-complementary break in the primer sequence which will replace the original sequence (Figure 1).
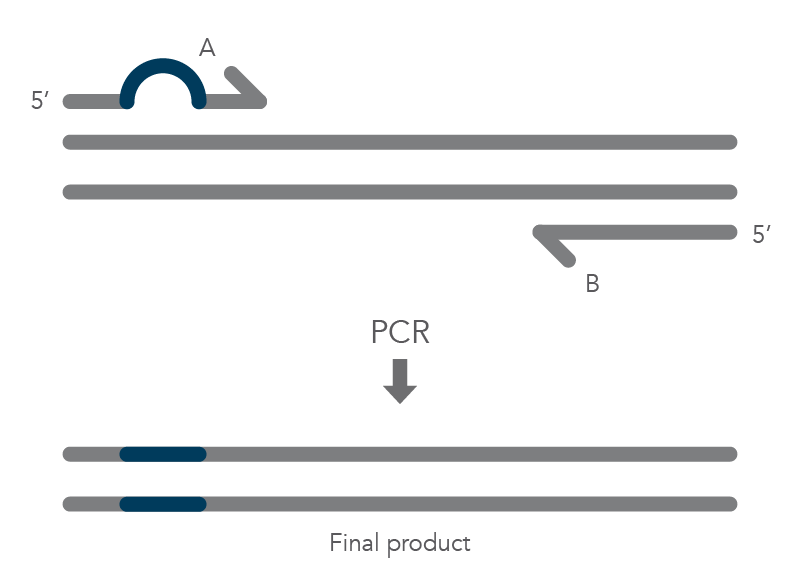
The PCR protocol for terminal additions involves adding to the sequence at the 5’ end of primer B, which is used with the complementary primer A for amplification of the new PCR product that contains the terminal additions (Figure 2).
Primer Extension Mutagenesis
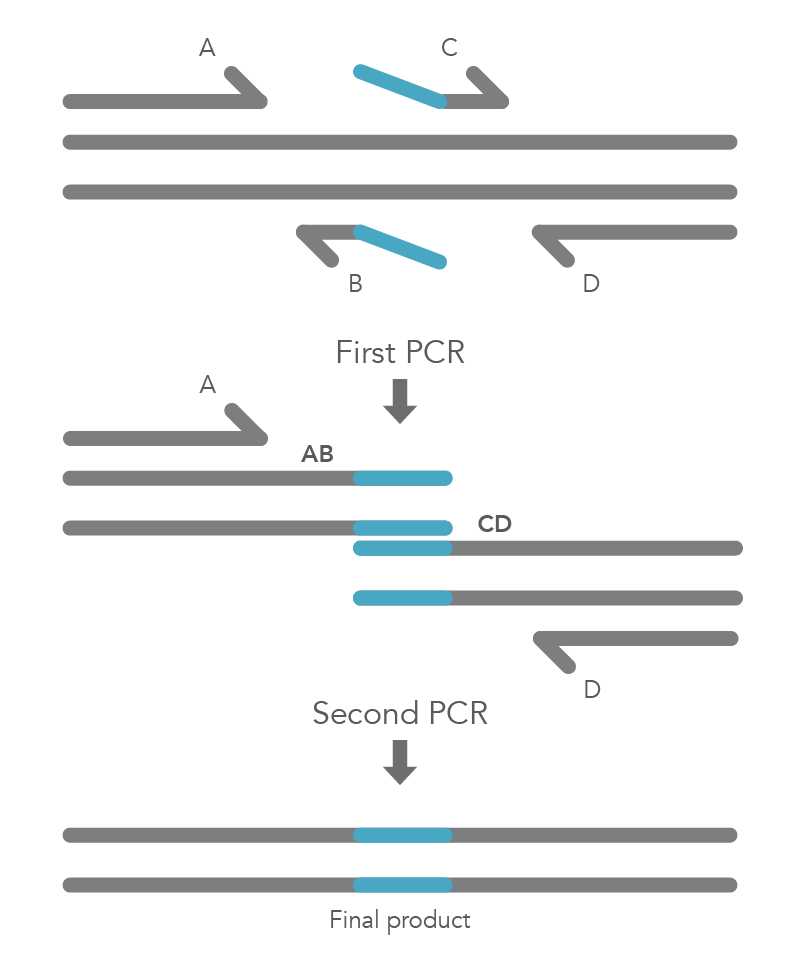
Site-directed mutagenesis by primer extension involves incorporating mutagenic primers in independent, nested PCRs before combining them in the final product [2]. The reaction requires flanking primers (B and C) to be positioned on either side of the region to be deleted, so that this region does not become part of the AB and CD fragments. The B and C primers include 5’ tail sequences that are complementary to the segment of target DNA upstream of the deletion for primer C or downstream of the deletion for primer B. The final two PCR amplicons have complementary overlapping regions so that the AB fragment will hybridize to the CD fragment in the second PCR reaction. Longer insertions can be incorporated by using especially long primers, such as IDT Ultramer™ oligonucleotides. Since the wild-type sequence is not amplified, the final AD product will not contain the deleted fragment (Figure 4).
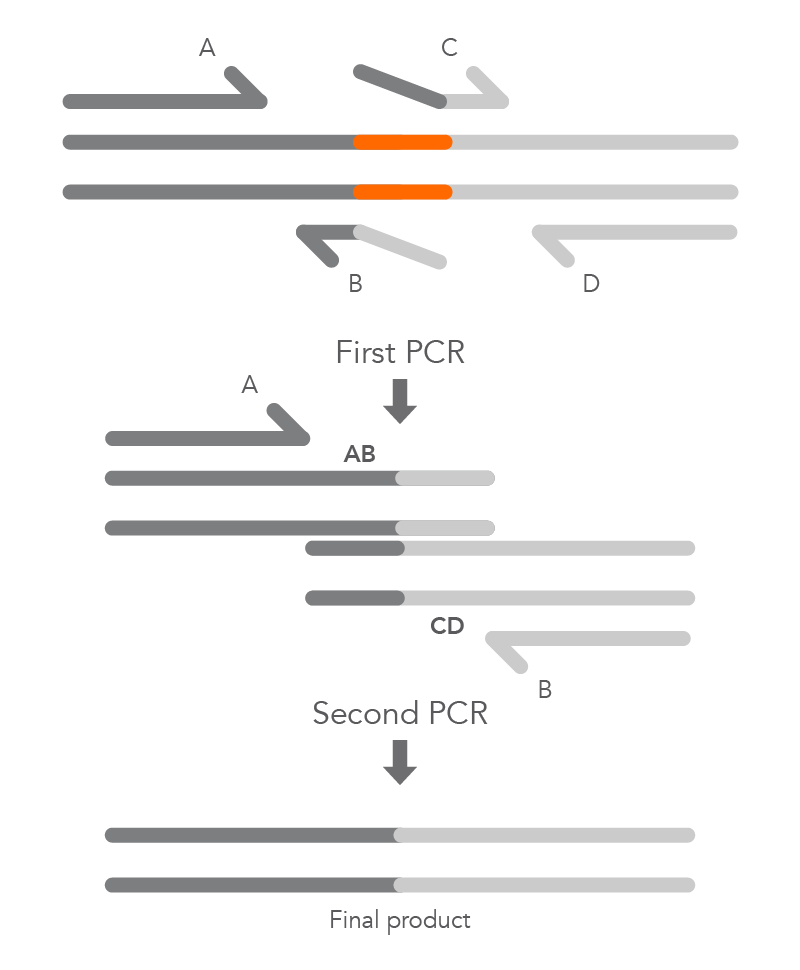
To create a deletion, the internal primers, B and C, are positioned at either side of the region to be deleted to prevent it from being incorporated within fragments AB and CD from the first round of PCR. The complementary sequences at the ends of these fragments, created by primers B and C, enable hybridization of AB to CD during the second round of PCR, and the final product with the desired deletion (AD) is created (Figure 5).
Inverse PCR Mutagenesis
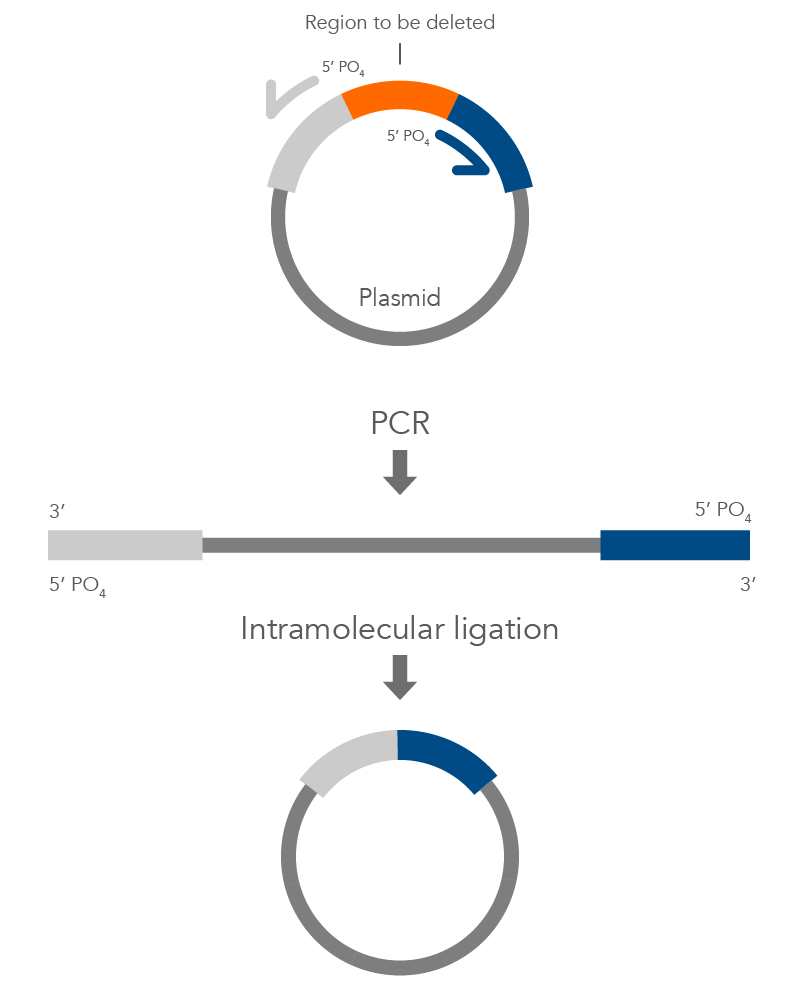
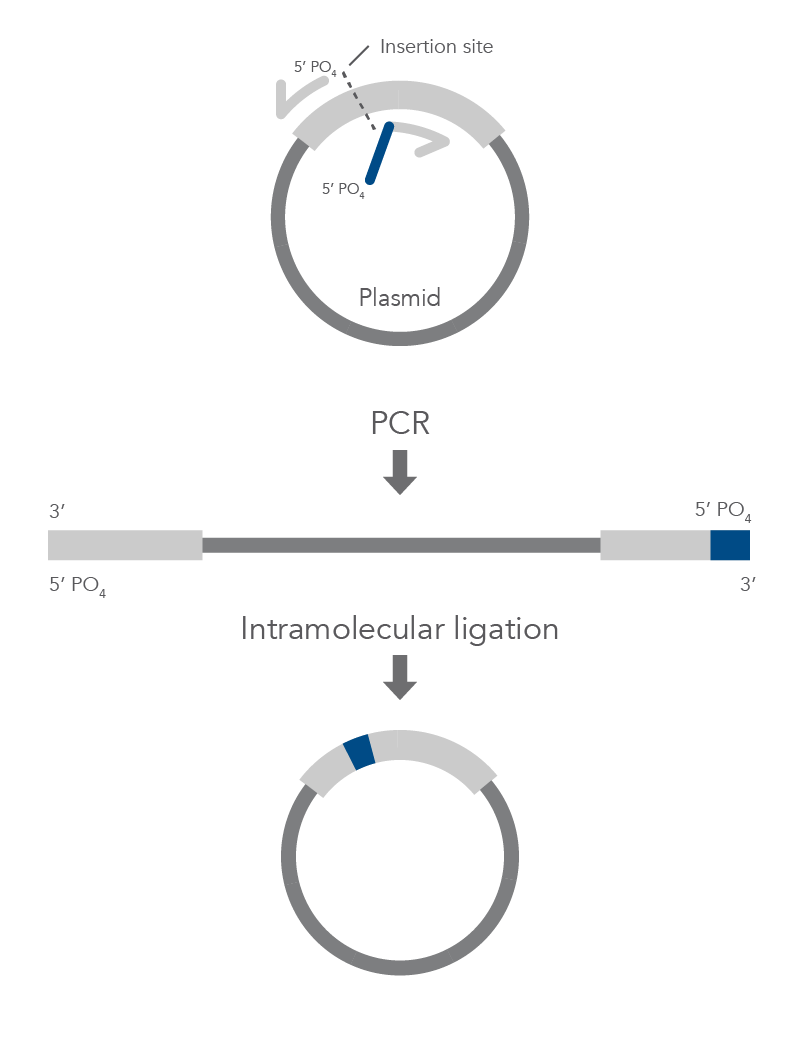
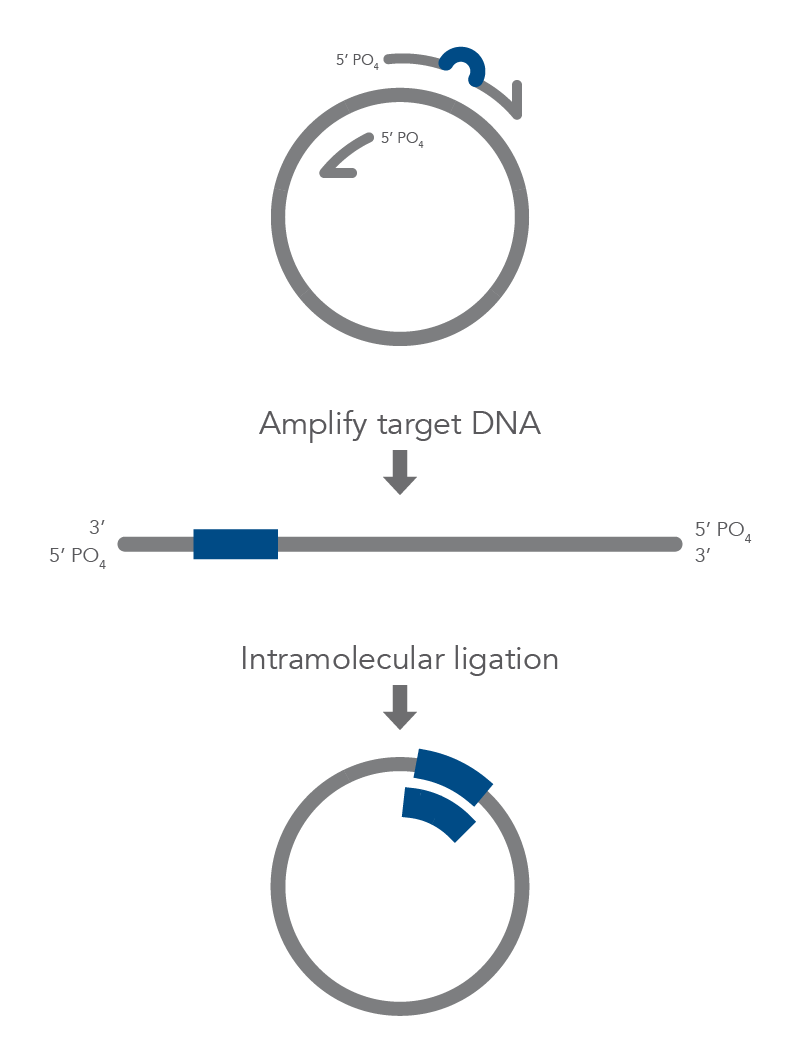
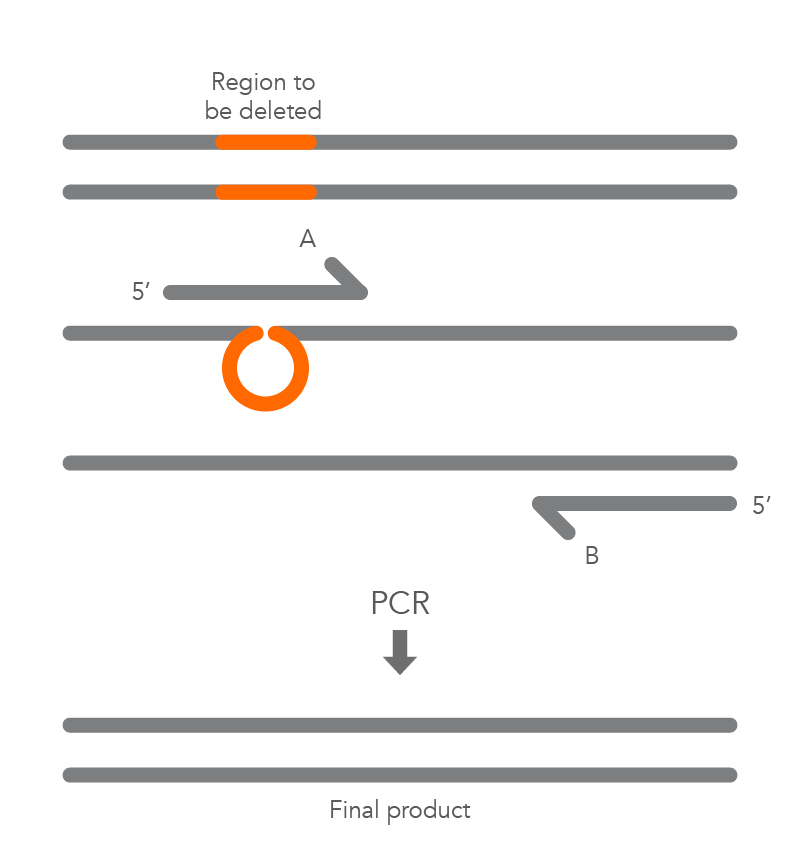
Inverse PCR enables amplification of a region of unknown sequence using primers oriented in the reverse direction [3]. An adaptation of this method can be used to introduce mutations in previously cloned sequences. Using primers incorporating the desired change, an entire circular plasmid is amplified to delete (Figure 6), substitute (Figure 7), or insert (Figure 8) the desired sequence.